TUS Kardiyoloji Çalışma Rehberi
EKG, ritim, göğüs ağrısı ve akut kardiyoloji yaklaşımını tek merkezde toplayan Akademi sayfası.
Pediatri, Nöroloji, Dermatoloji ve Patoloji'nin ortak kesişim kümesi olan Nörokutan Sendromları (Fakomatozlar) mekanizmaları ve patognomonik bulgularıyla öğrenin. NF1, Tuberoz Skleroz, VHL ve Sturg...
Admin · TUS hazırlığını sistemli hale getirmek için hazırlanmış eğitim içeriği.
Bu yazı daha geniş bir içerik kümesinin parçası. Aynı konuya bağlı seçilmiş yazıları merkez sayfadan takip edebilirsiniz.
EKG, ritim, göğüs ağrısı ve akut kardiyoloji yaklaşımını tek merkezde toplayan Akademi sayfası.
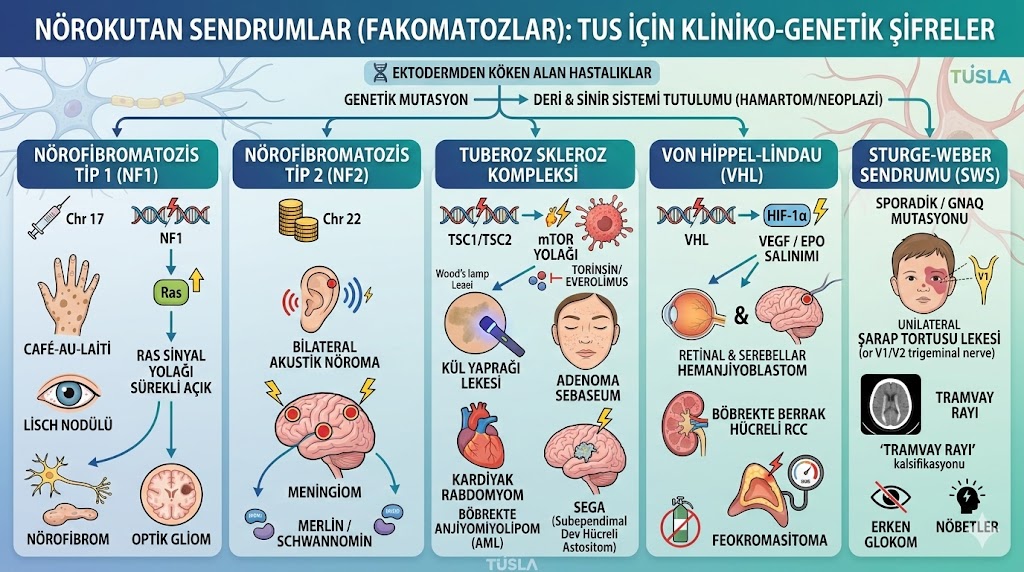
Pediatri, Nöroloji, Dermatoloji ve Patoloji'nin ortak kesişim kümesi olan Nörokutan Sendromları (Fakomatozlar) mekanizmaları ve patognomonik bulgularıyla öğrenin. NF1, Tuberoz Skleroz, VHL ve Sturg...
Yazı içindeki ana bölümlere hızlıca geçmek için aşağıdaki bağlantıları kullanabilirsiniz.
Merhaba değerli meslektaşım,
TUS hazırlık sürecinde öyle konular vardır ki, tek bir hastalığın fizyopatolojisini anladığınızda Pediatri, Nöroloji, Dermatoloji, Patoloji ve hatta Üroloji testlerinde bile soru yakalarsınız. Adayların genellikle gen isimleri ve cilt bulguları arasında kaybolduğu Nörokutan Sendromlar (Fakomatozlar), tam anlamıyla sınavın "multidisipliner gizli hazinesi" ve derece yaptıran spesifik alanlarından biridir.
Embriyolojik olarak hem sinir sisteminin hem de derinin ektodermden (ve kısmen nöral krestten) köken alması, bu dokulardaki genetik bir mutasyonun neden her iki sistemde de eşzamanlı lezyonlar (hamartomlar, neoplaziler) yarattığını çok iyi açıklar.
İnternetteki klasik "şunda şu leke var, bunda bu tümör var" şeklindeki yüzeysel listeleri unutun. TUS komitesi artık hücre içi sinyal yollarını (mTOR, HIF-1 $\alpha$) ve "en sık" görülen spesifik organ tutulumlarını sorguluyor. Şimdi bu devasa konuyu, ezberden uzaklaştırıp TUSLA stratejisiyle mekanizmalarına ayıralım.
> ### 💡 TUSLA Spot Bilgi / Özet Kutu
>
>
> Nörokutan sendromların geneli Otozomal Dominant (OD) geçer. İSTİSNA: Sturge-Weber Sendromu (Sporadiktir, genetik aktarılmaz!).
> Kromozom 17: Nörofibromatozis Tip 1 (NF1).
> Kromozom 22: Nörofibromatozis Tip 2 (NF2).
> Bilateral Akustik Nöroma (Vestibüler Schwannom) = Nörofibromatozis Tip 2.
> Kardiyak Rabdomyom (kendiliğinden gerileyebilen kalp tümörü) = Tuberoz Skleroz.
> Böbrekte Anjiyomiyolipom (AML) = Tuberoz Skleroz; Böbrekte Berrak Hücreli Karsinom (RCC) = Von Hippel-Lindau.
> Şarap tortusu lekesi (Port-wine stain) + Glokom + Tramvay rayı kalsifikasyon = Sturge-Weber Sendromu.
>
>
---
TUS'ta NF1 ve NF2 ayrımını yapmak çok kritiktir. Basitçe düşünün: NF1 daha çok "dışarıyı" (deri, periferik sinir, göz), NF2 ise "içeriyi" (santral sinir sistemi tümörleri) tutar.
A. Nörofibromatozis Tip 1 (Von Recklinghausen Hastalığı)
B. Nörofibromatozis Tip 2
Tuberoz Skleroz, vücudun hemen her organında hamartom (bulunduğu dokunun normal hücrelerinden oluşan ama düzensiz büyüyen kitle) üreten bir fabrikaya dönüşmesi durumudur.
VHL, anjiyogenezin (yeni damar oluşumu) şalterinin kapatılamadığı bir hastalıktır.
Bu hastalık genetik geçişli değildir. Somatik mozaizm dediğimiz, anne karnında gelişirken sonradan ortaya çıkan GNAQ gen mutasyonuna bağlıdır. Bu detay TUS'ta "Aşağıdakilerden hangisi kalıtsal değildir?" sorusunun cevabıdır.
---
Sınav kitapçığını açtığınızda, uzun bir vaka paragrafını saniyeler içinde çözmenizi sağlayacak o altın tablo:
| Hastalık | Genetik Geçiş / Kromozom | Deri / Göz Bulgusu (Patognomonik İpucu) | Karakteristik Tümör veya Organ Tutulumu |
| --- | --- | --- | --- |
| Nörofibromatozis Tip 1 | OD / Kromozom 17 | Café-au-lait lekesi, Lisch nodülü (İris) | Nörofibrom, Optik Gliom, Feokromasitoma |
| Nörofibromatozis Tip 2 | OD / Kromozom 22 | (Spesifik cilt bulgusu yok), Gençte katarakt | Bilateral Akustik Nöroma, Meningiom |
| Tuberoz Skleroz | OD / TSC1-TSC2 (mTOR) | Kül yaprağı lekesi, Adenoma sebaseum | Kardiyak Rabdomyom, Böbrekte AML, SEGA |
| Von Hippel-Lindau | OD / Kromozom 3 (VHL) | (Spesifik cilt bulgusu yok) | Serebellar/Retinal Hemanjiyoblastom, Böbrekte RCC |
| Sturge-Weber | SPORADİK (Kalıtsal Değil) | Şarap tortusu lekesi, Erken Glokom | Leptomeningeal anjiyom, Tramvay rayı kalsifikasyon |
---
Gördüğünüz gibi, Nörokutan Sendromlar birbirine benzeyen ezber kırıntıları değil; genetik bir şalterin bozulmasıyla ortaya çıkan, son derece mantıklı ve öngörülebilir klinik sendromlardır. "Böbrekteki kitlenin cinsi" veya "Lekenin Wood lambasındaki rengi" gibi detaylar, sizi 5 çeldirici şıktan doğrudan doğru cevaba götüren birer pusuladır.
Bu mekanizmaları zihninize kazıdığımıza göre, şimdi bu sendromların TUS vaka sorularında (özellikle karmaşık Pediatrik Nöroloji vakalarında) nasıl gizlendiğini test etmek ister misiniz? Hemen TUSLA platformu üzerinden "Pediatri - Nöroloji ve Genetik" soru çözüm kampına geçiş yapın, çıkmış ve çıkması muhtemel vaka şifrelerini benimle birlikte çözün!
Okumayı bitirdikten sonra seni bir sonraki mantıklı adıma taşıyan seçilmiş bağlantılar.
Bu konuda bağlantılı içerikleri ve yüksek getirili başlıkları tek merkezde gör.
Aynı konu kümesinde ilerlemek için seçilmiş ilgili Akademi yazısını aç.
Aynı konu kümesinde ilerlemek için seçilmiş ilgili Akademi yazısını aç.
Okuduğun karar mantığını soru çözümüyle pekiştir.
Aynı konu kümesinde ilerlemek için seçilmiş ilgili Akademi yazıları.